
Alexandr Gnezdilov Light Painting
Niema Moshiri, University of California San Diego
When you hear the term “evolutionary tree,” you may think of Charles Darwin and the study of the relationships between different species over the span of millions of years.
While the concept of an “evolutionary tree” originated in Darwin’s “On the Origin of Species,” one can apply this concept to anything that evolves, including viruses. Scientists can study the evolution of SARS-CoV-2 to learn more about how the genes of the virus function. It is also useful to make inferences about the spread of the virus around the world, and what type of vaccine may be most effective.
I am a bioinformatician who studies the relationships between epidemics and viral evolution, and I am among the many researchers now studying the evolution of SARS-CoV-2 because it can help researchers and public health officials track the spread of the virus over time. What we are finding is that the SARS-CoV-2 virus appears to be mutating more slowly than the seasonal flu which may allow scientists to develop a vaccine.
Cambridge University Library
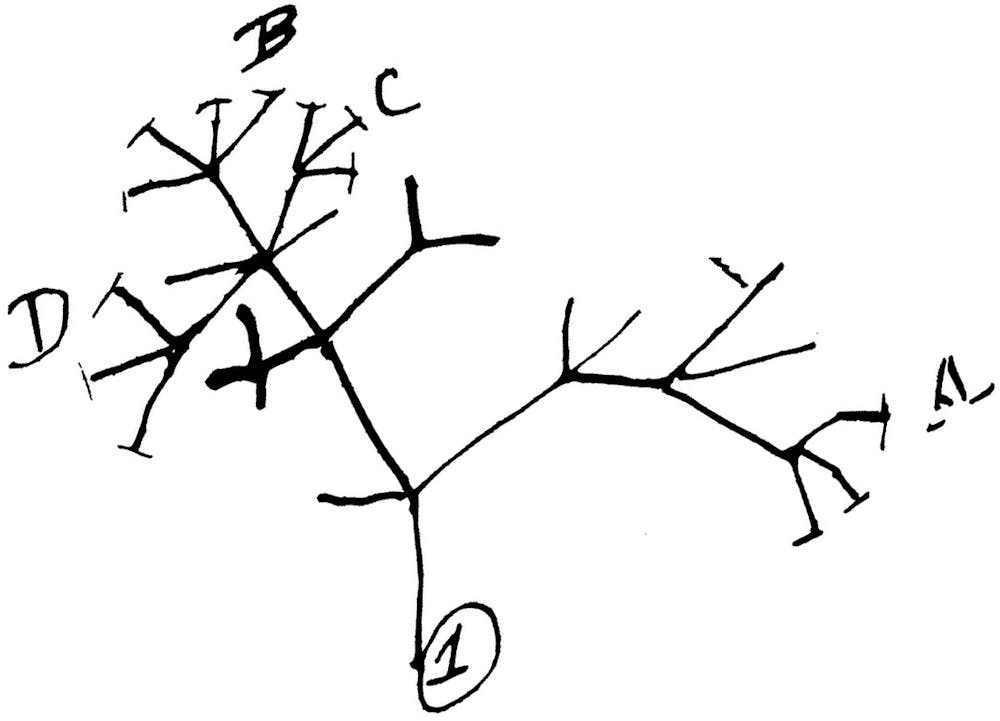
How do sequences evolve?
Viruses evolve by mutating. That is, there are changes in their genetic code over time. The way it happens is a little like that game of telephone. Amy is the first player, and her word is “CAT.” She whispers her word to Ben, who accidentally hears “MAT.” Ben whispers his word to Carlos, who hears “MAD.” As the game of telephone goes on, the word will transform further and further away from its original form.
We can think of a biological genetic material as a sequence of letters, and over time, sequences mutate: The letters of the sequence can change. Scientists have developed various models of sequence evolution to help them study how mutations occur over time.
Much like our game of telephone, the genome sequence of the SARS-CoV-2 virus changes over time: Mutations occur randomly, and any changes that occur in a given virus will be inherited by all copies of the next generation. Then, much as we could try to decode how “CAT” became “MAD,” scientists can use models on genetic evolution to try to determine the most likely evolutionary history of the virus.
How can we apply this to viruses like COVID-19?
DNA sequencing is the process of experimentally finding the sequence of nucleotides (A, C, G and T) – the chemical building blocks of genes – of a piece of DNA. DNA sequencing is largely used to study human diseases and genetics, but in recent years, sequencing has become a routine part of viral point of care, and as sequencing becomes cheaper and cheaper, viral sequencing will become even more frequent as time progresses.
RNA is a molecule similar to DNA, and it is essentially a temporary copy of a short segment of DNA. Specifically, in the central dogma of biology, DNA is transcribed into RNA. SARS-CoV-2 is an RNA virus, meaning our DNA sequencing technologies cannot directly decode its sequence. However, scientists can first reverse transcribe the RNA of the virus into complementary DNA (or cDNA), which can then be sequenced.
Given a collection of viral genome sequences, we can use our models of sequence evolution to predict the virus’s history, and we can use this to answer questions like, “How fast do mutations occur?” or “Where in the genome do mutations occur?” Knowing which genes are mutating frequently can be useful in drug design.
Tracking how viruses have changed in a location can also answer questions like, “How many separate outbreaks exist in my community?” This type of information can help public health officials contain the spread of the virus.
For COVID-19, there has been a global initiative to share viral genomes with all scientists. Given a collection of sequences with sample dates, scientists can infer the evolutionary history of the samples in real-time and use the information to infer the history of transmissions.
One such initiative is Nextstrain, an open-source project that provides users real-time reports of the spread of seasonal influenza, Ebola and many other infectious diseases. Most recently, it has been spearheading the evolutionary tracking of COVID-19 by providing a real-time analysis as well as a situation report meant to be readable by the general public. Further, the project enables the global population to benefit from its efforts by translating the situation report to many other languages.
As the amount of available information grows, scientists need faster tools to be able to crunch the numbers. My lab at UC San Diego, in collaboration with the System Energy Efficiency (SEE) Lab led by Professor Tajana Šimunić Rosing, is working to create new algorithms, software tools and computer hardware to make the real-time analysis of the COVID-19 epidemic more feasible.

from nextstrain.org/ncov
What have we learned about the epidemic?
Based on current data, it seems as though SARS-CoV-2 mutates much more slowly than the seasonal flu. Specifically, SARS-CoV-2 seems to have a mutation rate of less than 25 mutations per year, whereas the seasonal flu has a mutation rate of almost 50 mutations per year.
Given that the SARS-CoV-2 genome is almost twice as large as the seasonal flu genome, it seems as though the seasonal flu mutates roughly four times as fast as SARS-CoV-2. The fact that the seasonal flu mutates so quickly is precisely why it is able to evade our vaccines, so the significantly slower mutation rate of SARS-CoV-2 gives us hope for the potential development of effective long-lasting vaccines against the virus.
[Get facts about coronavirus and the latest research. Sign up for The Conversation’s newsletter.]![]()
Niema Moshiri, Assistant Teaching Professor of Computer Science & Engineering, University of California San Diego
This article is republished from The Conversation under a Creative Commons license. Read the original article.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}